Phylogenetics
Phylogenetic analysis models evolutionary relationships between different organisms, such as pathogens, based on genetic information. In the case of HIV, viral sequencing is integrated in routine care in resource-rich settings due to monitoring of drug resistance mutations. Hence, HIV viral sequences are available for many people with HIV, allowing to perform large-scale phylogenetic analyses. These analyses can complement mathematical modelling approaches by providing insights into transmission patterns beyond laws pre-defined by the model, such as super-spreading events or the occurrence of growing transmission clusters.
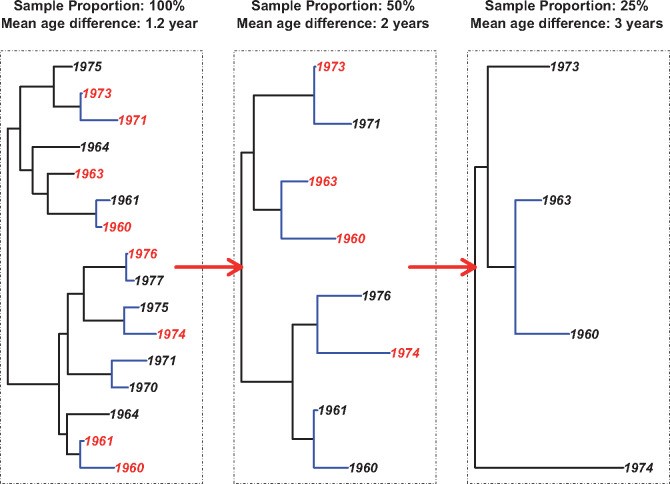
One of our group’s focus are phylogenetic analyses to understand transmission dynamics of the HIV epidemic in Europe. One publication to highlight would be to understand the impact of sample density on results concerning age patterns: “Inferring the age difference in HIV transmission pairs by applying phylogenetic methods on the HIV transmission network of the Swiss HIV Cohort Study”. The representativeness of the SHCS resistance database, which covers at least 27 per cent of the whole Swiss HIV epidemic, allowed us to analyze the impact of the sample proportion and the distance threshold on the age difference of observed pairs in the phylogenetic tree (see Figure). Both factors proved to influence the mean age difference of HIV transmission pairs, which was measured by the absolute difference in birth years. The age difference decreased almost monotonically both with a stricter, i.e., smaller, distance threshold, and a higher sample proportion. Especially for low sample proportions, deriving the age difference of pairs in the phylogenetic tree, or similar quantitative measures, can be misleading and requires extensive sensitivity analysis.
Figure: Schematic presentation of the down-sampling process
Additional projects include the following:
- Phylogenetics and molecular evolution to understand and curb the HIV pandemic
- Similar But Different: Integrated Phylogenetic Analysis of Austrian and Swiss HIV-1 Sequences Reveal Differences in Transmission Patterns of the Local HIV-1 Epidemics
- Identifying and Characterizing Trans Women in the Swiss HIV Cohort Study as an Epidemiologically Distinct Risk Group
- Using longitudinally sampled viral nucleotide sequences to characterize the drivers of HIV-1 transmission
- Differences in Social and Mental Well-Being of Long-Term Survivors among People who Inject Drugs and Other Participants in the Swiss HIV Cohort Study: 1980–2018